
Name of Authors: Isaac Filella
Name of Institution: Barcelona Supercomputing Center
e-mail: isaac.filella1@bsc.es
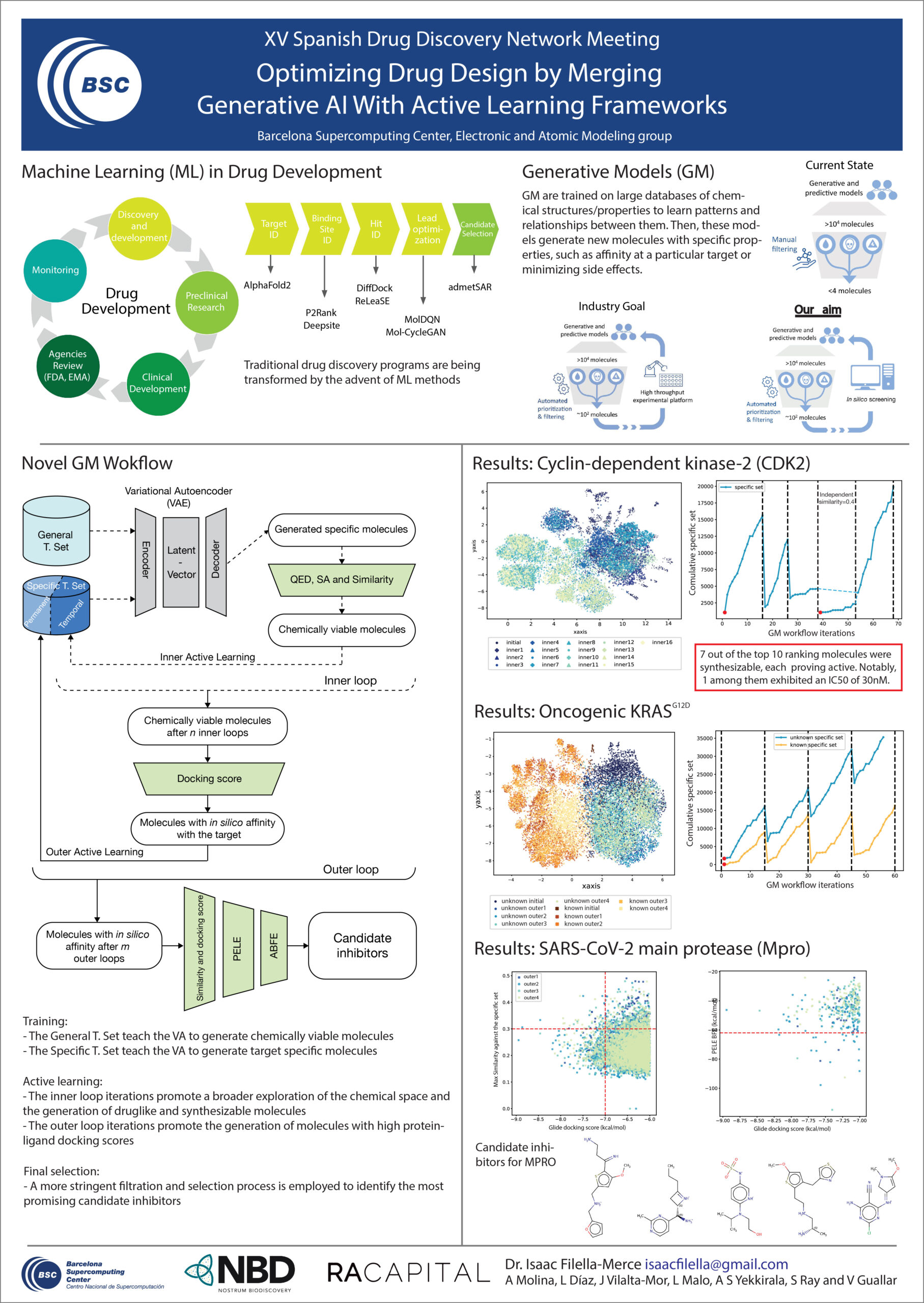
Traditional drug discovery programs are being transformed by the advent of machine learning methods. Among these, Generative AI methods (GM) have gained attention due to their ability to design new molecules and enhance specific properties of existing ones. However, current GM methods have limitations, such as low affinity towards the target, unknown ADME/PK properties, or the lack of synthetic tractability. To improve the applicability domain of GM methods, we have developed a workflow based on a variational autoencoder coupled with active learning steps. The designed GM workflow iteratively learns from molecular metrics, including drug likeliness, synthesizability, similarity, and docking scores. In addition, we also included a hierarchical set of criteria based on advanced molecular modeling simulations during a final selection step. We tested our GM workflow on two model systems, CDK2 and KRAS. In both cases, our model generated chemically viable molecules with a high predicted affinity toward the targets. Particularly, the proportion of high-affinity molecules inferred by our GM workflow was significantly greater than that in the training data. Notably, we also uncovered novel scaffolds significantly dissimilar to those known for each target. These results highlight the potential of our GM workflow to explore novel chemical space for specific targets, thereby opening up new possibilities for drug discovery endeavors.
{kind=link}